Research Focus
Ethos Research & Development conducts novel research into the neurobiological processes which drive the development and worsening of chronic pain disorders. Our research aims to identify and validate novel pain biomarkers that allow healthcare providers to diagnose and treat patients based on underlying pathophysiological mechanisms rather than symptomology. Mechanistic biomarkers of pain will not only improve our understanding of pain biochemistry and allow for truly personalized treatment strategies but will also pave the way for the development of novel, non-opioid pain drugs.
Functional Biomarkers of Pain
The Functional Biomarkers of Pain test panel was developed by Ethos Research & Development in 2015 and is available to healthcare providers across the United States through our industry partner, Ethos Laboratories (Newport, KY). This novel, pain-specific biomarker test panel evaluates urinary markers of cytokine mediated inflammation, oxidative stress, micronutrient status and neurotransmitter turnover, all of which are known to play a role in the development and/or worsening of chronic pain disorders. Individual biomarkers in this unique test panel were selected based on several criteria, including but not limited to:
- Relevance: Every biomarker in this test panel has a direct link to the development and/or worsening of pain. What does this mean? Abnormal results indicate a possible cause or underlying explanation for a patient’s pain.
- Prevalence: Biomarkers reveal commonly perturbed pathways in the chronic pain population. What does this mean? We are not testing for rare genetic disorders. Our metabolomic biomarkers assist with identifying common causes of pain.
- Treatability: Every biomarker in this test panel can be modulated or corrected if found to be abnormal. What does this mean? We are identifying underlying, TREATABLE causes of pain. Most importantly, indicated treatments for most abnormal biomarker results are safe, cost effective, often over the counter (OTC), and non-opioid.
Examples of Functional Pain Biomarkers
Methylmalonic acid
Methylmalonic acid (also known as MMA or Methylmalonate) is a sensitive and highly specific functional marker for the assessment of Vitamin B12 (cobalamin) status. Vitamin B12 status is an especially important consideration for pain practitioners as deficiencies of this essential vitamin can cause demyelination of nerves leading to painful neuropathies, axonal death and subacute combined degeneration of the spinal cord (1). Low levels of Vitamin B12 have been linked to polyneuropathy, trigeminal neuralgia, migraine, depression, mania, optic nerve atrophy, neuropsychiatric disorders, and various functional disabilities (2-6). Deficiencies in vitamin B12 can also result in elevated levels of homocysteine in the blood and urine due to its crucial role as a cofactor in amino acid metabolism. Elevated levels of homocysteine have been recognized as an independent risk factor for cardiovascular disease and stroke. An early diagnosis of vitamin B12 deficiency is crucial as response to treatment is dependent on the extent of nerve damage and the timing of replacement therapy (7).
The importance of screening chronic pain patients for vitamin B12 deficiency cannot be overstated due to the prevalence of malabsorption leading to deficiency. Other risk factors for Vitamin B12 deficiency include the use of medications such as proton pump inhibitors (PPIs), H2-receptor antagonists, metformin, colchicine, cholestyramine, and frequent or long-term use of anticonvulsants and/or antibiotics (3). Gastric surgery, intestinal bacterial overgrowth, hypothyroidism, diabetes and aging represent other risk factors for vitamin B12 deficiency. Due to the large number of medications (both prescription and over-the-counter), medical conditions and lifestyle choices that can precipitate vitamin B12 deficiency, combined with the ease, cost and effectiveness of replacement therapy, widespread screening is recommended within the chronic pain population. In addition to replenishing Vitamin B12 stores, treatment with various forms of cobalamin has been shown to provide pain relief, alleviate pain behaviors, improve nerve conduction and exert neuronal protection by promoting regeneration of injured nerves and antagonizing glutamate-induced neurotoxicity (8-13).
Evaluating levels of Methylmalonic acid is the most sensitive and specific method for assessing vitamin B12 status. Normalized urinary Methylmalonic acid levels greater than or equal to 2.3 μg/mg of creatinine are indicative of a functional vitamin B12 deficiency. Individuals with levels in the ‘high normal range’ should be monitored and retested or instructed to begin supplementation in order to decrease the likelihood of them becoming deficient. Replacement therapy for vitamin B12 deficiency is safe, convenient and cost effective. Options include oral, sublingual and/or intramuscular formulations. If malabsorption (opposed to inadequate intake of animal products) is suspected to be the precipitating factor for the deficiency, intramuscular injections will likely be the most effective formulation for replenishment.
Xanthurenic acid
Xanthurenate (also known as Xanthurenic acid) is a sensitive marker of Vitamin B6 (pyridoxine) status. It is a byproduct of the hepatic kynurenine pathway that is strongly dependent on adequate pyridoxal phosphate, the active form of vitamin B6. Vitamin B6 is an essential vitamin required for the synthesis of proteins (including neurotransmitters such as serotonin and norepinephrine), the formation and integrity of the nerve insulating myelin sheath, the production of anti-inflammatory mediators and immune system function. A deficiency of vitamin B6 can cause peripheral neuropathy, migraine, chronic pain, depression, seizures and other neuropsychiatric disorders (14-16). Due to its central role in nerve health and function, an optimal level of vitamin B6 is necessary for successful and prolonged pain management and screening for deficiencies should be part of a laboratory based pain workup. While the direct impact of vitamin B6 deficiency on nerve health and function has been recognized for decades, recent research indicates that inflammation and the inflammatory process may actually drive a tissue-specific depletion of vitamin B6. Investigators conclude that the low circulating levels of vitamin B6 commonly seen in patients with inflammatory disease may result from the removal of vitamin B6 from circulation to meet the higher demand within certain tissues during the inflammatory process (17). This critical link between the inflammatory process and vitamin B6 status may explain why circulating B6 levels appear to exhibit an inverse correlation with the degree of pain in patient populations. Evaluating levels of Xanthurenate is a sensitive and selective method for assessing vitamin B6 status. The metabolic conversion of Xanthurenate along the kynurenine pathway is heavily dependent on vitamin B6 such that a deficiency or insufficiency will manifest as an accumulation of Xanthurenate. In addition to its role as a marker of vitamin B6 status, recent research indicates that elevated levels of Xanthurenate, and therefore vitamin B6 insufficiency, may play a central role in the development of insulin resistance and its progression to type 2 diabetes (18, 19).
Pyroglutamic acid
Elevated levels of Pyroglutamate indicate glutathione depletion which renders nerve cells susceptible to oxidative damage. Glutathione is the most important and abundant intracellular antioxidant in all aerobic cells (20). Its powerful antioxidant effects are due to its dual roles as a scavenger of reactive oxygen species (ROS) and reactive nitrogen species (RNS) and as a substrate for the detoxifying enzymes glutathione peroxidase (GPx) and GSSG reductase (GR) (21). Glutathione represents the most integral component of our natural antioxidant defense system and optimal levels are critical for combatting oxidative stress and ensuring cell survival. Oxidative stress is the direct consequence of an increased generation of free radicals and/or reduced physiological activity of our antioxidant defense system. The central role of oxidative stress in the development, maintenance and worsening of peripheral neuropathy is widely recognized and more recent studies have even identified free radicals as key players in the production of pain and the lowering of local nociceptor thresholds leading to hyperalgesia (22-24). Studies have also illustrated a direct link between levels of oxidative stress and the resulting susceptibility of skeletal muscle to fatigue and pain (22). As the body of literature describing the role of oxidative stress in chronic pain conditions continues to grow the capacity to maintain glutathione responses becomes a more crucial factor for the successful and prolonged management of pain. This is especially true for patients who take regular doses of acetaminophen as the toxic metabolite of acetaminophen, N-acetyl-p-benzoquinone imine (NAPQI) is detoxified by glutathione. In the absence of sufficient levels of glutathione, NAPQI will combine with structural proteins leading to toxicity and hepatic damage. Not only does acetaminophen directly deplete glutathione through conjugation reactions but its metabolism and excretion also depletes glycine and the sulfur-containing amino acids that are required for glutathione synthesis (25). The depletion of glutathione by NAPQI and the consumption of necessary sulfated amino acids during acetaminophen metabolism is widely under recognized as a source of glutathione depletion and its relevance to pain management cannot be overstated. Evaluating glutathione status and replenishing deficiencies in order to decrease the susceptibility of cells to further oxidative damage can be achieved safely and cost effectively. Of the several interventions for improving glutathione capacity, one of the most highly effective strategies is combined oral N-acetyl-L-cysteine (NAC) augmented by taurine to spare the activity of that synthetic pathway along with glycine (another easily-depleted glutathione precursor) (25, 26).
Kynurenine Pathway metabolites
Quinolinate and Kynurenate are neuroactive metabolites of the kynurenine pathway which is significantly upregulated in response to inflammation. As a result elevated levels of Quinolinate and Kynurenate serve as sensitive markers of chronic, systemic inflammation and the upregulation of this critical pathway has been shown to play a central role in the comorbidity of pain and depression (27). Up regulation of the kynurenine pathway in response to inflammation, stress or chronic activation of the innate immune system impacts the development and severity of pain via two main mechanisms: Firstly, up regulation of this pathway results in a decreased production of serotonin as both pathways utilize tryptophan as a substrate. Decreased levels of serotonin not only lead to depression but also diminish the activity of descending inhibitory pain pathways which under normal serotonin supply act to inhibit pain (28-33). Secondly, up regulation of the kynurenine pathway increases circulating levels of Quinolinic acid which contributes to heightened nociception and an increased susceptibility to neurotoxicity via its interaction with glutamate receptors (34). Quinolinic acid is therefore not only a sensitive marker of systemic inflammation but also a bioactive modulator of pain perception due to its action on NMDA receptors linked to nociceptor systems. Kynurenate is an NMDA receptor antagonist that is also elevated during episodes of inflammation or chronic stress but appears to play a neuroprotective role by antagonizing the neurotoxic effects of Quinolinate. Therefore, the relative concentrations of these two kynurenine metabolites provides an indication of the potential risk for neuronal degeneration. As the QUIN/KYN ratio increases so too does the risk of nerve cell death as a result of systemic inflammatory disease. In the absence of inflammation, Quinolinate will not be elevated and high Kynurenate reinforces abnormal Xanthurenate due to a B6 deficient impact on the hepatic kynurenine pathway. Magnesium and glycine are frequently helpful for antagonizing glutamatergic NMDA receptors, offsetting the Quinolinate agonistic effects.
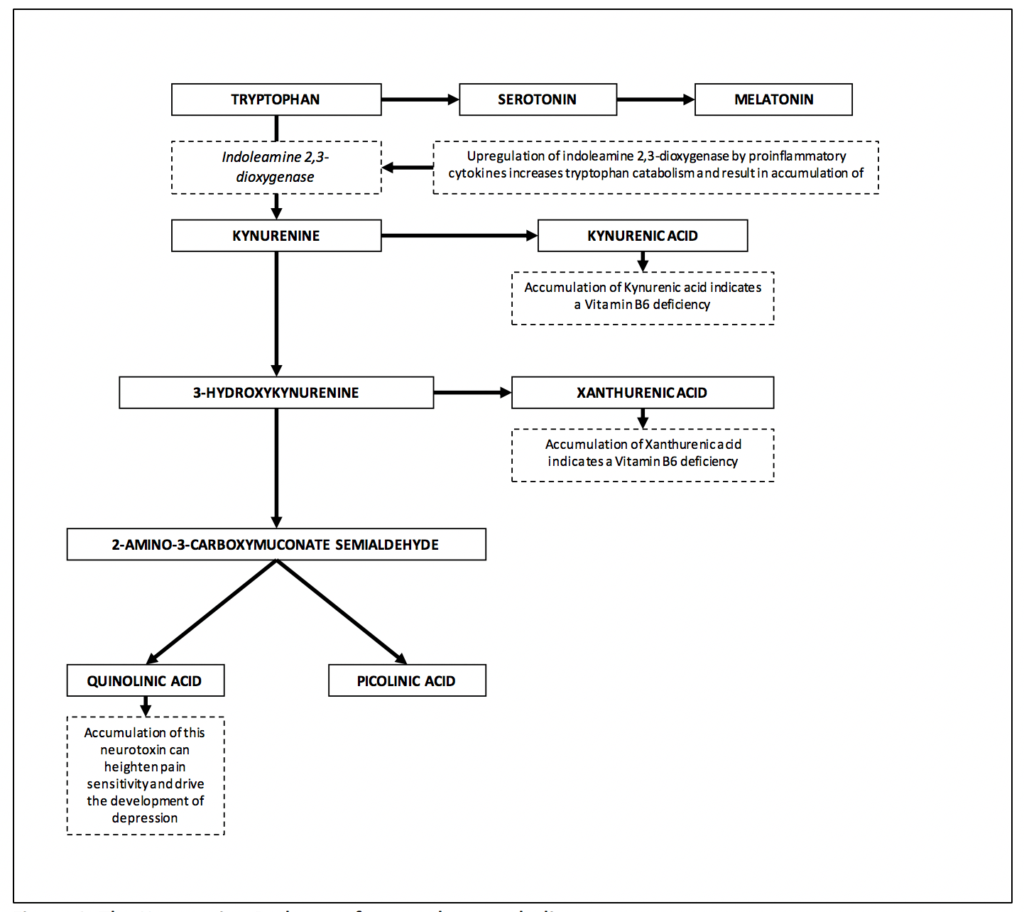
Figure 1. The Kynurenine Pathway of tryptophan catabolism.
5-hydroxyindoleacetic acid (5-HIAA)
5-Hydroxyindoleacetate (5-HIAA) is the primary metabolite of serotonin which, along with norepinephrine is the principle mediator of descending inhibitory pathways which act to suppress pain. It is produced by synaptic monoamine oxidase action on serotonin, and SSRI therapies increase those losses due their inhibition of reuptake that increases serotonin synaptic dwell times. Abnormally low levels of 5-HIAA can indicate inadequate production of serotonin which has been linked to depression and is believed to contribute to hyperactive pain processing (29). Serotonin is a monoamine neurotransmitter which is biochemically derived from dietary tryptophan but during episodes of inflammation, tryptophan is preferentially metabolized along the kynurenine pathway leading to decreased serotonin production. This inflammation induced ‘shunt’ away from serotonin and melatonin production results in greater circulating levels of neuroactive, brain-specific kynurenine metabolites such as Quinolinate and decreased levels of serotonin and melatonin. It is the deregulation of this pathway that provides the mechanistic link between systemic inflammation and diseases associated with low serotonin status such as chronic pain and depression. Patients with low levels of 5-HIA may benefit from serotonin precursor therapies such as 5-hydroxytryptophan or L-tryptophan. Elevated levels of 5-HIA are commonly observed in patients who are taking serotonin reuptake inhibitor medications such as SSRI’s or SSNRI’s.
Vanilmandelate
Vanilmandelate is the urinary metabolite of norepinephrine, which along with serotonin is the principal mediator of endogenous pain inhibiting systems know as descending inhibitory pathways. Optimal levels of norepinephrine are required for the activation of descending inhibitory pathways which act to inhibit pain. Deficient levels of this neurotransmitter system are believed to contribute to hyperactive pain processing while elevated levels of norepinephrine signify chronic stress and can lead to the generalized hypervigilant state often observed in patients with fibromyalgia (29, 35). Norepinephrine is a unique biomarker of pain as it can have a marked impact on the development, maintenance and perception of pain when present at sub optimal or elevated levels. Sufficient levels are required to activate descending inhibitory pathways which act to suppress ascending pain signals at the spinal level but an overproduction of norepinephrine can heighten ones sensitivity to pain as a result of generalized hypervigilance and perceptual amplification. While urinary levels of Vanilmandelate do not reflect or directly correlate to levels in the CNS, they do reflect overall rates of biosynthesis and breakdown and provide physicians with a non-invasive means to identify patients who may benefit from therapies designed to modulate the production of catecholamines.
Retrospective Analysis of Functional Biomarker Results in a Chronic Pain Population
In 2019, Ethos Laboratories conducted a large, retrospective analysis of Functional Biomarker test results from 17,833 unique patients whose samples were submitted to and analyzed by Ethos Laboratories. The aim of the analysis was to determine which biochemical abnormalities are most commonly detected in long term opioid users.
Results of Retrospective Analysis
77% of chronic pain patients exhibited at least one abnormal biomarker result (n=13,765). The most common abnormal biomarker finding was elevated Quinolinic acid, which was observed in 29% of patients (n=5107). Elevated Pyroglutamate, indicative of glutathione depletion, was observed in 19% of patients (n=3314). Elevated Xanthurenic acid, indicative of Vitamin B6 insufficiency, was observed in 17% of patients (3025). Elevated levels of the acrolein metabolite 3-hydroxypropyl mercapturic acid (3-HPMA) were observed in 21% of patients (n=3667). Elevated Methylmalonic acid (MMA), indicative of a Vitamin B12 deficiency, was observed in 10% of patients (n=1827) while abnormally low levels of neurotransmitter metabolites were observed in 8% of patients (n=1456).
Read more about these important findings here.
Publications
-
Nassan FL, Gunn JA, Hill MM, Coull BA, Hauser R. High phthalate exposure increased urinary concentrations of quinolinic acid, implicated in the pathogenesis of neurological disorders: Is this a potential missing link? Environ Res. 2019; 172:430-436.
-
Gunn JA, Hill MM, Cotten B, Deer TR. An analysis of biomarkers in chronic pain patients. In press.
-
Deer TR, Gunn J. Blood testing in chronic pain management. Pain Physician. 2015; 18(2):E157-161.
References
-
Babior BM, Bunn HF. Megaloblastic anemias. In: Braunwald E, Fauci AS, Kasper DL, Hauser SL, Longo DL, Jameson JL, editors. Harrison’s principles of internal medicine, 15th ed. New York: McGraw-Hill; 2001. p. 674-680.
-
Sun AL, Ni YH, Li XB, Zhuang XH, Liu YT, Liu XH, Chen SH. Urinary Methylmalonic acid as an indicator of early vitamin B12 deficiency and its role in polyneuropathy in type 2 diabetes. Journal of Diabetes Research. 2014; Epub 2014 Feb 26.
-
Briani C, Torre CD, Citton V, Manara R, Pompanin S, Binotto G, Adami F. Cobalamin deficiency: clinical picture and radiological findings. Nutrients. 2013; 5:4521-4539.
-
Aggarwal A, Wood I. Low vitamin B12 syndrome in trigeminal neuralgia. J Pain Relief. 2012; 1(5).
-
Ipcioglu OM, Ozcan O, Gultepe M, Tekeli H, Senol MG. Functional vitamin B12 deficiency represented by elevated urine Methylmalonic acid levels in patients with migraine. Turk J Med Sci. 2008; 38(5):409-414.
-
Oberlin BS, Tangney CC, Gustashaw KAR, Rasmussen HE. Vitamin B12 deficiency in relation to functional disabilities. Nutrients. 2013; 5:4462-4475.
-
Huang CR, Chang WN, Tsai NW, Lu CH. Serial nerve conduction studies in vitamin B12 deficiency-associated polyneuropathy. Neurol Sci. 2011; 32:183-186.
-
Kikuchi M, Kashii S, Honda Y, Tamura Y, Kaneda K, Akaike A. Protective effects of methylcobalamin, a vitamin B12 analog, against glutamate-induced neurotoxicity in retinal cell culture. Investigative ophthalmology and visual science. 1997; 38(5):848-854.
-
Kong X, Sun X, Zhang J. The protective role of mecobalamin following optic nerve crush in adult rats. Yan Ke Xue Bao. 1997; 20(3):171-177.
-
Akaike A, Tamura Y, Sato Y, Yokota T. Protective effects of a vitamin B12 analog, methylcobalamin, against glutamate cytotoxicity in cultured cortical neurons. European Journal of Pharmacology. 1993; 241(1):1-6.
-
Devathasan G, Teo WL, Mylvaganam A. Methylcobalamin in chronic diabetic neuropathy: a double-blind clinical and electrophysiological study. Clinical Trials Journal. 1986; 23(2):130-140.
-
Kuwabara S, Nakazawa R, Azuma N et al. Intravenous methylcobalamin treatment for uremic and diabetic neuropathy in chronic hemodialysis patients. Internal Medicine. 1999; 38(6):472-475.
-
Ishihara H, Yoneda M, Yamamoto W. Efficacy of intravenous administration of methylcobalamin for diabetic peripheral neuropathy. Med Consult N Remedies. 1992; 29(1):1720-1725.
-
Levenson JL. Textbook of psychosomatic medicine, 2nd ed. Arlington, VA: American Psychiatric Publishing, 2011:517.
-
Hammond N, Wang Y, Dimachkie M, Barohn R. Nutritional neuropathies. Neurol Clin. 2013; 31(2):477-489
-
Beer MH, Porter RS, Jones TV, Kaplan JL, Berkwits M, eds. The Merck Manual, 18th ed. Rahway, NJ: Merck Research Laboratories, 2006:26-61, 1036-1039, 1261-1263.
-
Chiang EP, Smith DE, Selhub J, Dallal G, Wang YC, Roubenoff R. Inflammation causes tissue-specific depletion of vitamin B6. Arthritis Res Ther. 2005; 7(6):R1254-R1262.
-
Oxenkrug GF. Increased plasma levels of Xanthurenic acid and Kynurenic acids in type 2 diabetes. Mol Neurobiol. 2015; 52(2):805-810.
-
Oxenkrug GF, Ratner R, Summergrad P. Kynurenines and vitamin B6: link between diabetes and depression. J Bioinform Diabetes. 2013; 1(1):1-12.
-
Chesney JA, Eaton JW, Mahoney J. Bacterial Glutathione: a sacrificial defense against chlorine compounds. J Bacteriol. 1996; 178(7):2131-2135.
-
Rizzardini M, Lupi M, Bernasconi S, Mangolini A, Cantoni L. Mitochondrial dysfunction and death in motor neurons exposed to the glutathione-depleting agent ethacrynic acid. Journal of the Neurological Sciences. 2003; 207:51-58.
-
Vecchiet J, et al. Relationship between musculoskeletal symptoms and blood markers of oxidative stress in patients with chronic fatigue syndrome. Neuroscience Letters. 2003; 335:151-154.
-
Khalil Z, Liu T, Helme RD. Free radicals contribute to the reduction in peripheral vascular responses and the maintenance of thermal hyperalgesia in rats with chronic constriction injury. Pain. 1999; 79:31-37.
-
Luo ZD, Cizkova D. The role of nitric oxide in nociception. Curr Rev Pain. 2000; 4(6):459-466.
-
Emmett M. Acetaminophen toxicity and 5-oxoproline (Pyroglutamic acid): a tale of two cycles, one an ATP-depleting futile cycle and the other a useful cycle. Clin J Am Soc Nephrol. 2014; 9(1):191-200.
-
Metges CC, Yu YM, Cai W, Lu XM, Wong S, et al. Oxoproline kinetics and oxoproline urinary excretion during glycine- or sulfur amino acid-free diets in humans. Am J Physiol Endocrinol Metab. 2000; 278(5):E868-876.
-
Kim H, et al. Brain Indoleamine 2,3-dioxygenase contributes to the comorbidity of pain and depression. The Journal of Clinical Investigation. 2012; 122(8):2940-2954.
-
Iyengar S, Webster AA, Hemrick-Luecke SK, Xu JY, Simmons RM. Efficacy of duloxetine, a potent and balanced serotonin-norepinephrine reuptake inhibitor in persistent pain models in rats. J Pharmacol Exp Ther. 2004; 311:576-584.
-
Marks DM, Shah MJ, Patkar AA, Masand PS, Park GY, Pae CU. Serotonin-norepinephrine reuptake inhibitors for pain control: premise and promise. Curr Neuropharmacol. 2009; 7(4): 331-336.
-
Miura H, Ozaki N, Sawada M, Isobe K, Ohta T, Nagatsu T. A link between stress and depression: shifts in the balance between kynurenine and serotonin pathways of tryptophan metabolism and the etiology and pathophysiology of depression. Stress. 2008; 11(3):198-209.
-
Myint AM, Kim YK. Cytokine-serotonin interaction through IDO: a neurodegenerative hypothesis of depression. Med Hypotheses. 2003; 61(5-6):519-525.
-
Mico JA, Gibert-Rahola J, Casas J, Rojas O, Serrano MI, Serrano JS. Implication of beta 1 and beta 2-adrenergic receptors in the antinociceptive effect of tricyclic antidepressants. Eur Neuropsychopharmacol. 1997; 7(2): 139-145.
-
Sawynok J, Esser MJ, Reid AR. Antidepressants as analgesics: an overview of central and peripheral mechanisms of action. J Psychiatry Neurosci. 2001; 26(1):21-29.
-
Stone TW, Perkins MN. Quinolinic acid: a potent endogenous excitant at amino acid receptors in CNS. Eur J Pharmacol. 1981; 72(4):411-412.
-
McDermid AJ, Rollman GB, McCain GA. Generalized hypervigilance in fibromyalgia: evidence of perceptual amplification. Pain. 1996; 66(2-3):133-144.